公众号/
编辑 | 萝卜皮

密度泛函理论(DFT)的定理建立了多体系统的局部外部势与其电子密度、波函数以及单粒子约化密度矩阵之间的双射映射。
在此基础上,罗格斯大学(Rutgers University)和纽约大学(New York University)的研究人员证明基于单电子约化密度矩阵(reduced density matrices)的 机器学习 模型可用于生成替代电子结构方法。
该团队为从小分子(如水)到更复杂的化合物(如苯和丙醇)的系统生成局部和混合 DFT、Hartree-Fock 和完整构型相互作用理论的替代品。代理 模型 使用单电子约化密度矩阵作为要学习的中心量。根据预测的密度矩阵,研究人员表明标准量子化学或第二个机器学习模型可用于计算分子可观测值、能量和原子力。
代理模型基本上可以生成标准电子结构方法可以生成的任何内容,从带隙和 Kohn-Sham 轨道到节能的从头算分子动力学模拟和红外光谱;它解释了非谐性和热效应,而不需要采用计算成本高昂的算法。
该研究以「Machine learning electronic structure methods based on the one-electron reduced density matrix」为题,于 2023 年 10 月 7 日发布在《Nature Communications》。

通常使用计算模型来预测分子和材料特性,从而代替或在进行昂贵的实验之前进行预测。它们还用于解释实验观察背后的复杂电子和核动力学。当这些计算策略需要评估系统的电子结构时,它们往往成为计算瓶颈,延长求解时间。因此,一个重要且及时的目标是开发能够以降低的计算成本,甚至完全绕过电子结构计算来提供复杂系统的电子结构的方法。
机器学习方法的标准用途是针对单个预测的数量,这些数量是通过一些描述符来学习的。例如,电子能量的预测(包括应用「δ」学习概念的预测)、偶极矩和极化率等。这样的作案方式并不理想。如果目标是模型尚未训练过的量,则需要训练新模型来预测该量。一个相关的例子是红外 (IR) 光谱的计算,其中需要谱线位置和强度。学习分子系统或材料势能面的典型机器学习模型只能通过分子动力学模拟和速度自相关函数分析来预测谱线位置。然而,为了预测强度,需要偶极矩的自相关函数。
建立两个机器学习模型(一个用于能量表面,一个用于偶极矩)的通常过程非常耗时,但最终是可以避免的。值得注意的是,有各种正在开发和补充的技术,涉及使用对称 2 或 N 中心表示或深度神经网络表示来学习关于局域轨道的哈密顿矩阵。
理想的机器学习方法应该从其所包含的信息量密集的数量中进行学习。最通用、最理想的机器学习模型将学习多体电子波函数。根据波函数,研究人员可以预测势能面、偶极矩和任何其他感兴趣的量。多体波函数的 ML 模型与其他精确波函数求解器(例如量子蒙特卡罗)相比正在变得具有竞争力。
然而,计算和训练数据大小的复杂性阻碍了它们的广泛适用性。幸运的是,可以利用 DFT 和降密度矩阵泛函理论(RDMFT)的严格双射图将焦点从多体波函数转移到其他相关量。当考虑自旋系统时,1-rdm(the 1-electron reduced density matrix) 的 ML 模型需要额外的约束和功能。
电子密度可以根据局部原子中心描述符来学习。虽然使用这些方法获得的能量可以精确到 1 kcal ⋅ mol^−1 左右,但模型密度通常与目标存在百分之几的偏差。然而,即使有这些小偏差,模型密度也不能直接用于评估 DFT 算法中的离子力。这是因为自洽的电子密度必须收敛到更严格的阈值内,以进行精确的力计算。利用 Hohenberg 和 Kohn 定理,电子密度可以用作 ML 模型中的目标量,其中外部电势作为特征。这可以恢复模型电子密度,即使不完全准确,仍然可以成功地用作准确学习 DFT 和耦合簇等方法的能量和力的特征。
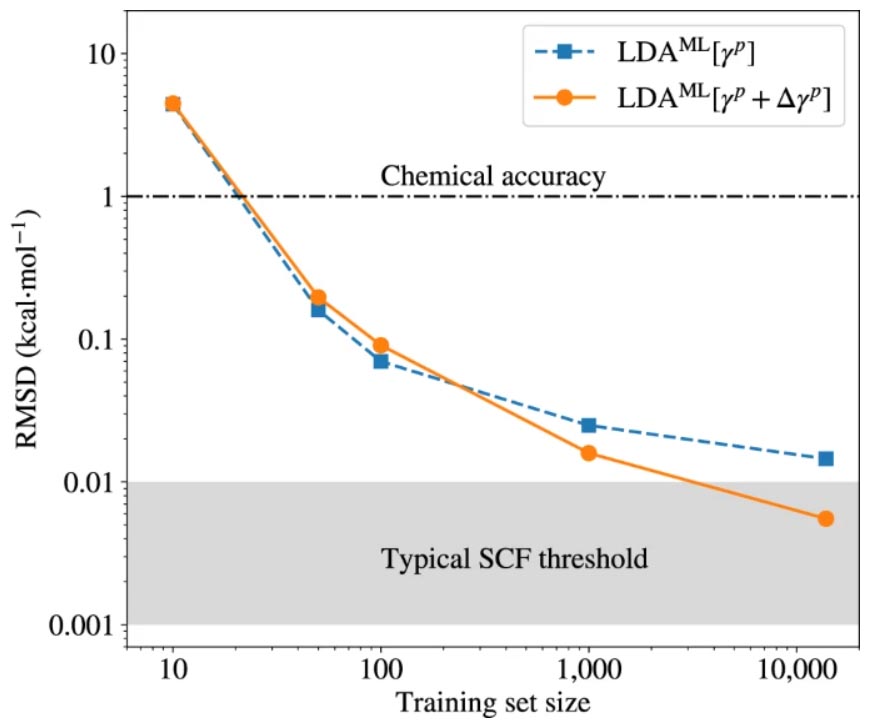
图示:替代电子结构方法的准确性作为苯分子训练集大小的函数。(来源:论文)
即使非局部相关性很重要,1-rdm 也正在成为描述量子系统的强大特征。在最新的研究中,罗格斯大学和纽约大学的研究人员接受了学习 1-rdm 的挑战,其准确度使得预测的 1-rdm 与标准电子结构软件提供的 1-rdm 基本上没有区别。
该团队通过在高斯型轨道 (GTO) 上表示外部势和目标 1-rdms 来实现这一目标,并设计一个高效的训练集生成器。这使他们能够提供「替代电子结构方法」,预测 1-rdms,然后提供有用的量(例如能量、力、带隙、轨道),这些量与标准电子结构软件计算的量一样准确和有用。
该团队展示了水和苯等刚性分子以及 1- 和 2-丙醇等柔性分子的 DFT、HF 和后 HF 替代物。首先,他们的模型学习了这些系统的 1-rdm 作为外部势的函数。然后,从 DFT 和 RDMFT 机器学习严格的映射,从而预测 1-rdm 到能量和 1-rdm 到原子力映射。
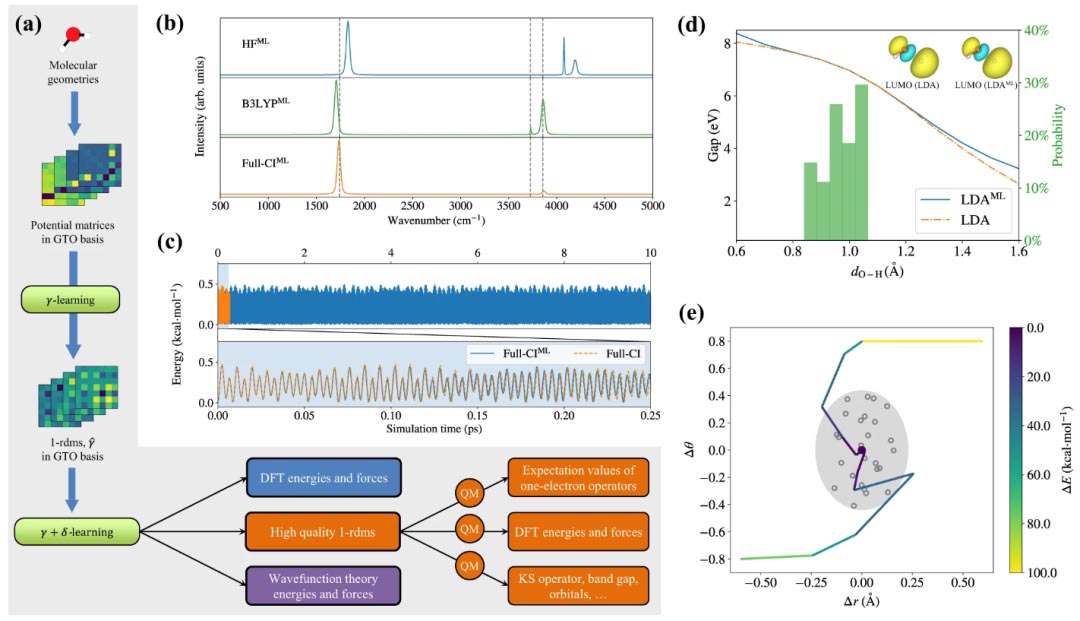
图示:水替代电子结构方法的性能。(来源:论文)
该团队的替代方法可以稳健地预测几何优化、从头算动力学和分子偶极矩的红外光谱。由于其多功能性,替代方法不仅可以预测结构和动力学,还可以预测单电子算子和 Kohn-Sham 轨道的期望值。他们预测了几个分子的 HOMO 和 LUMO 轨道以及能隙,即使对于远离训练集采样的构型空间的几何形状,结果也非常接近传统方法。
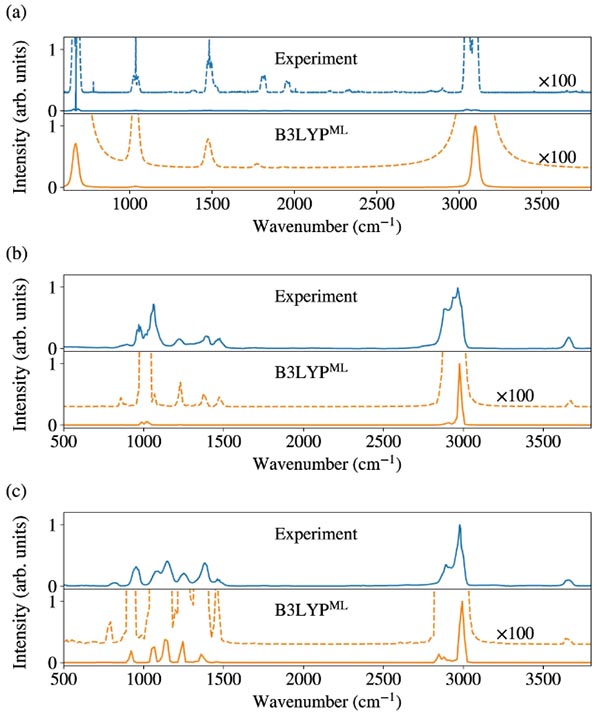
图示:用替代电子结构方法预测气相红外光谱。(来源:论文)
这项研究是一个概念证明,表明替代电子结构方法可以取代传统电子结构方法来完成大多数计算化学任务。然而,将该算法扩展到高阶 rdms 将能够计算双电子算子、能量和力的期望值,并将在未来考虑。凝聚相系统和更大的分子也是未来发展的目标。为了接近大分子,可以结合从小分子学到的单独分子片段的 1-rdms 来构建 1-rdms 的初始猜测。然后可以单独学习它们的耦合。
这项工作中开发的方法收集在全 Python QMLearn 软件中,该软件可以从 GitLab 免费获得,并可以通过 pip install qmlearn 轻松安装。
QMLearn 由以下类组成:(1)收集训练集的数据库;(2) QM 引擎(该团队使用 PySCF,尽管也支持其他引擎)能够在 GTO 上生成训练集和矩阵元素以及计算能量和原子力所需的基础设施;(3) 结构处理程序(该团队使用原子模拟环境)。ASE 用于处理分子几何形状,包括驱动分子动力学模拟;(4) ML 模块,例如 scikit-learn 或 Tensorflow(当前版本的 QMLearn 仅支持 scikit-learn)。
论文链接:https://www.nature.com/articles/s41467-023-41953-9