自监督学习蛋白质序列, 自然语言处理助力蛋白质工程新飞跃
公众号/将门创投
From:BAIR 编译:T.R
随着BERT等自然语言模型取得的突破性进展,人们逐渐认识到大模型可以在无标签数据上学习语言的强大表示。这些表示可以有效用于编码语义和句法。在自然语言处理成功的启发下,研究蛋白质的专家也在尝试着将自然语言处理的方法应用于蛋白质的结构预测中。
那么目前自然语言处理的前沿方法能够如何改善蛋白质预测任务呢?让我们来一探究竟。
无所不能的蛋白质
蛋白质对于生物的运转必不可少,从运输氧气的血红蛋白到人眼中的感光蛋白,从运输离子的输运蛋白到肌肉中的肌肉蛋白,它们的存在为生命造就多姿多彩的发展。理解蛋白质的结构和损坏机理不仅能够让我们对疾病的分子学机理有着更好的了解,更能帮助我们找到更好的方式对抗疾病!
蛋白质除了是维生的必要物质,更是生产各种抗体和疫苗的有效方式,同时还可以通过个性化改造让细菌具有分解废物的能力,生产出具有去污功效的酶。如果能够更深入地理解蛋白质,更多的新功能就可以被不断开发出来造福人类。
蛋白质的基础问题
蛋白质的本质是由一系列共价键衔接起来的氨基酸分子链。构建蛋白质的氨基酸有20种,通过的特定序列的空间构型形成了复杂的蛋白质结构。理论上这种离散的序列被称为蛋白质的一级结构。


图中展示了由丝氨酸、组氨酸和半胱氨酸分子构成的蛋白质片段表示。
但在细胞中,蛋白质是一种复杂的三维大分子结构,这种三维结构对于蛋白质生物特性的理解具有关键的作用。在更复杂的空间构型中,蛋白质的局部几何构型则被称为二级结构,使得蛋白质的不同区段表现出不同的行为特征。而整个蛋白质的全局结构则被称之为三级结构,它决定蛋白质的整体行为。

上图表示了蛋白质的一级、二级和三级结构层次。
对于一个特定的蛋白来说,它的氨基酸序列可以表示成下面的样子:

而研究的目标则在于从基础的序列中恢复出整个蛋白质的三维空间构型,也就是说需要确定每一个氨基酸分子的三维空间坐标,这对于蛋白质的生物功能十分重要,上面的序列对应的结构如下图所示:
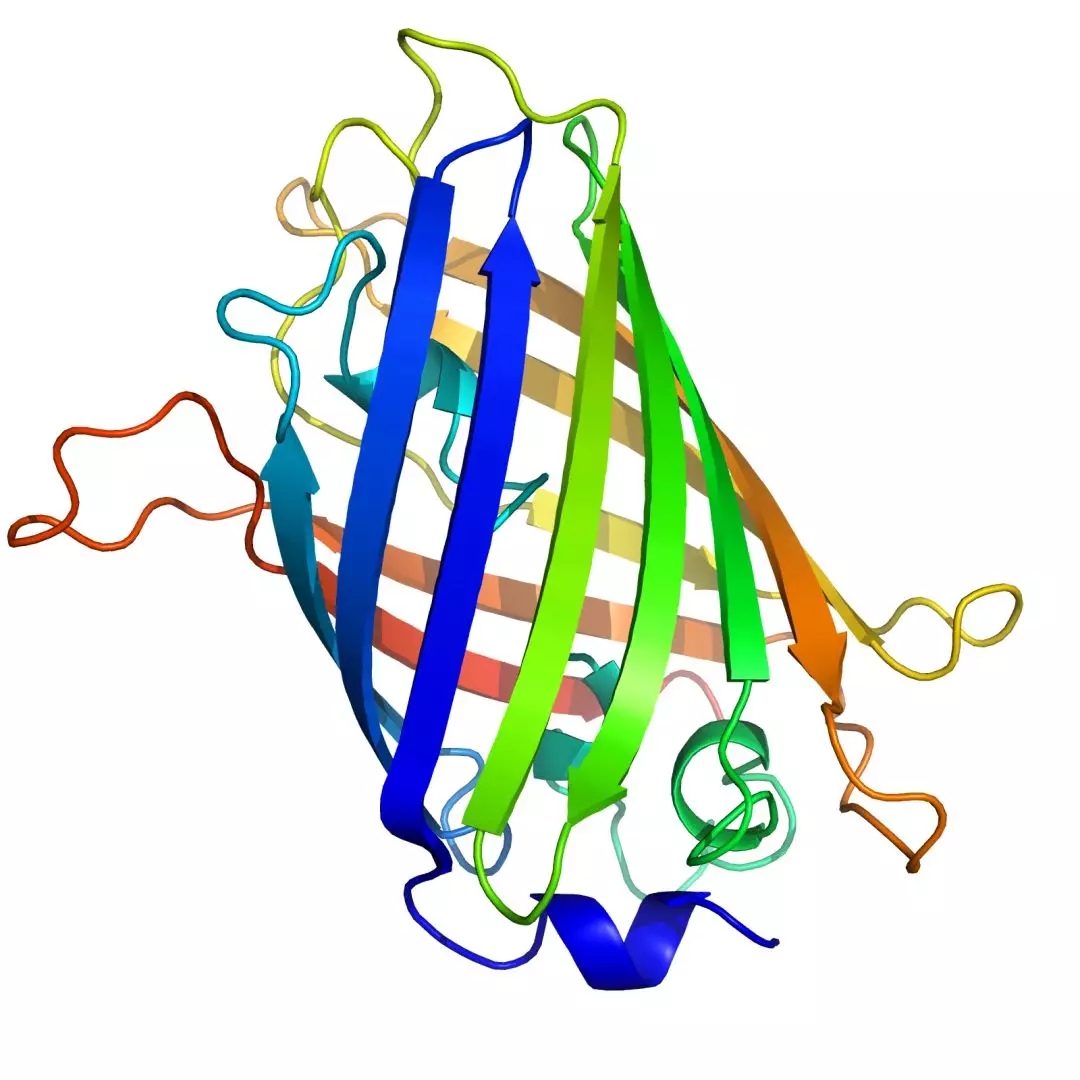
绿色荧光蛋白的空间结构
这是生物化学中最具挑战的问题,也是最为复杂的预测问题之一。
如果通过监督学习的方式来解决这一问题,我们就需要数据标签,而且是十分庞大的数据标签。针对蛋白质的空间构型预测,我们需要对蛋白质分子中的每一个氨基酸进行空间坐标位置标记。对一个蛋白质分子进行标记已经是很复杂的工作了,何况需要标记非常庞大的数量。人工标记的数据量远远落后于新蛋白质产生的速度。
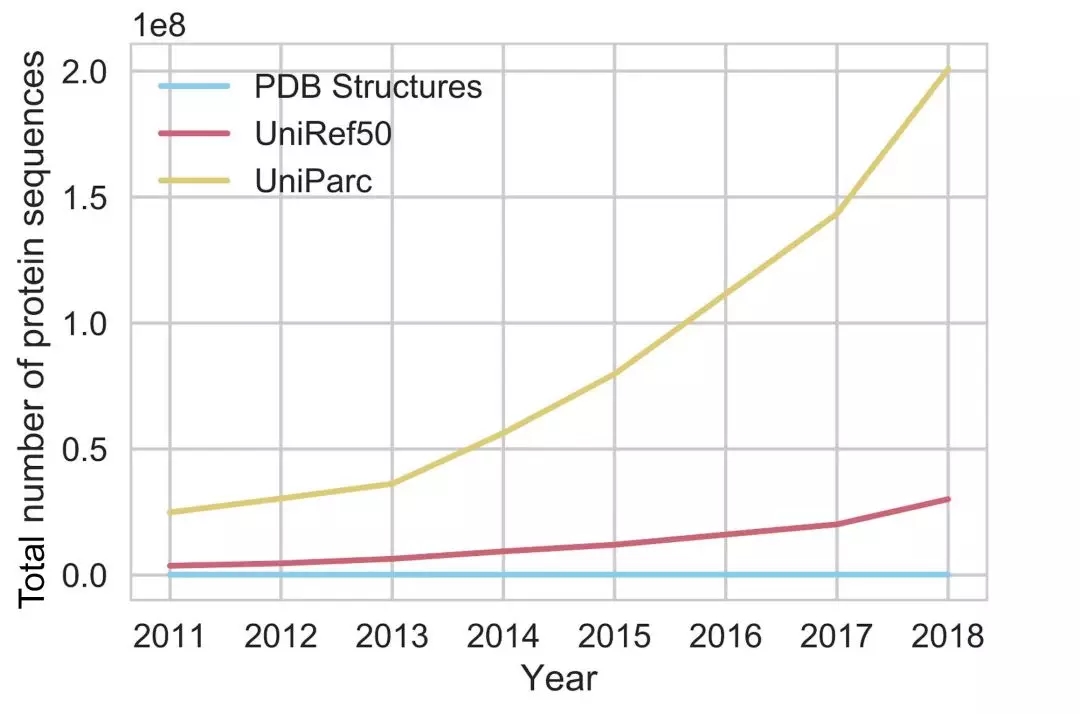
蛋白质的指数增长
那么除了监督学习我们还能如何解决这一问题呢?
拥抱无标签数据
虽然无法标记数量巨大的数据集,但是好消息是我们目前拥有庞大的无标签蛋白质序列数据集。如果我们可以从中抽取出有用的信息,这将为我们提供强有力的信息源。
目前使用这些无标签数据集的常用范式主要是序列比对方法。利用已知的蛋白质去数据库中进行扫描,找出与之近亲的序列。在最简单的情况下,如果我们找到了已知结构的近亲序列,可以将近亲结构的匹配位置映射到对应的已知序列上去。
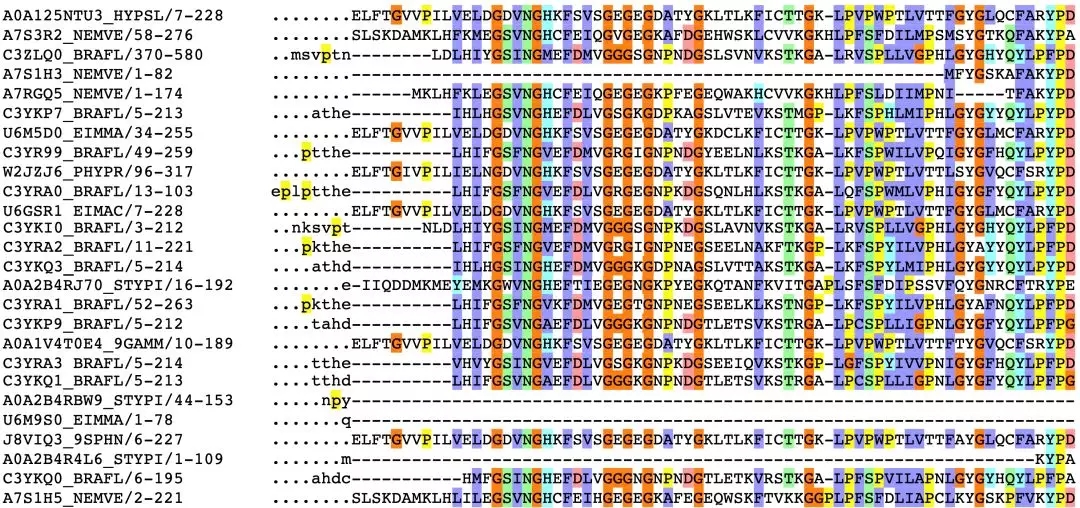
绿色荧光蛋白家族的部分序列比对。维多利亚管发光水母不是唯一一种能够产生荧光蛋白的的生物,还包括了 sea anenomes海葵, lancelets文昌鱼, and potato fungus马铃薯真菌.
上图中展示了不同的近亲序列结构,其中最左侧是序列ID,随后代表序列结构位置的表示,颜色表示不同的氨基酸族,紫色代表疏水氨基酸,红色代表带电氨基酸、绿色为不带电氨基酸。点和线代表了序列中的间隔位置。
氨基酸序列的进化关系意味着什么呢?进化本质上一种约束条件,如果某种改变造成了蛋白质结构损坏,生物也会因为失去蛋白质的对应功能而受损。所以研究人员希望从近亲序列中找到氨基酸中哪些进化位置是完全可以自由变化的,哪些位置是部分约束的,哪些位置是完全被约束的。
这种对于蛋白质家族的表示成为多重序列比对,是蛋白质结构预测中的关键输入。进化模型为蛋白质结构预测提供了良好的理论基础,但要如何有效的抽取序列特征呢?我们需要更强有力的工具!
通过自监督学习蛋白质的语言
这些生物学的中蛋白质序列概念让我们不禁想到了自然语言处理的方法,而且最近利用自监督预训练使得自然语言得到了巨大的突破。那么是不是有可能通过自然语言处理的方式来学习出蛋白质序列的表达,从而预测可能的蛋白质进化结构,为科研人员指明实验的方向,增强蛋白质的功能呢?
让我们来看一个自然语言处理的例子,如果数据集中有一个句子:
Let’s stick to improvisation in this skit
随机去除句子中的某些单词输入模型:
Let’s stick to [?] in this skit
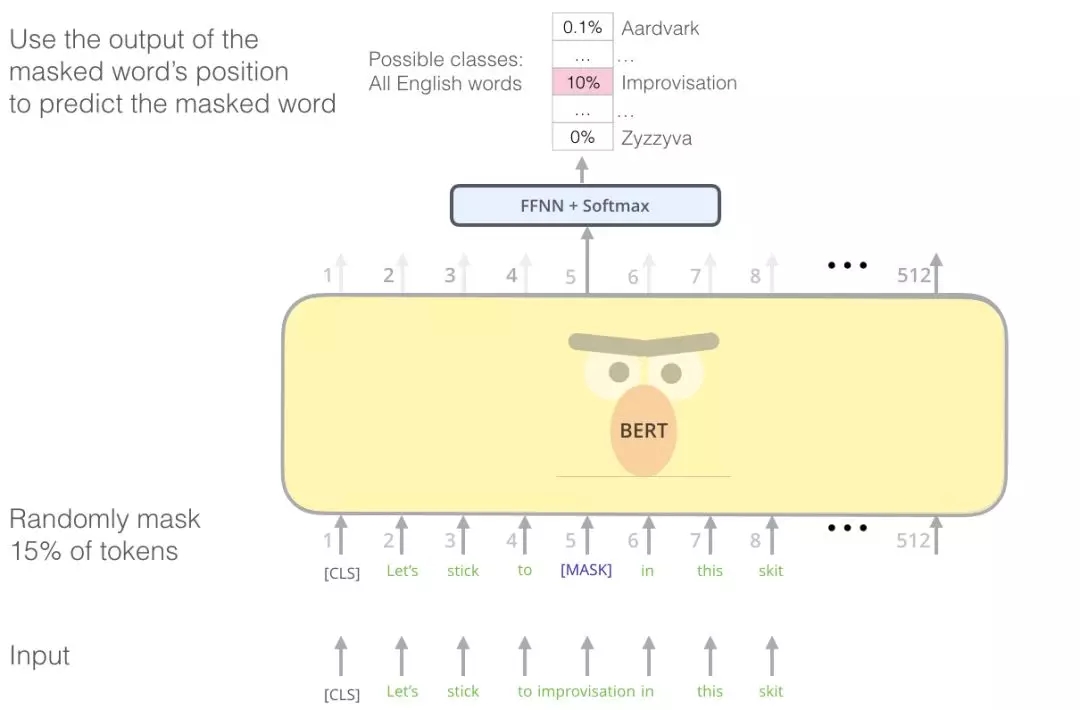
基于transformer的语言模型构建
模型将输出针对不同词汇输出概率,并利用交叉熵来惩罚预测错误词汇的模型。在观察了大量的句子序列后,像谷歌BERT和OpenAI的GPT2等模型就能学习到序列的有效特征,并为后续任务提供语言的有效表达。
我们可以将同样的方法应用到蛋白质序列上,并在氨基酸的水平进行序列特征抽取,下面是绿色荧光蛋白的序列
MSKGEELFTGVVPILVELDGDVNGHKFSVS…
随机去除某些位置后,让模型预测出这些未知位置:
MSKGE?LFT?VVP?ILVELDGDV?GHKFSVS…
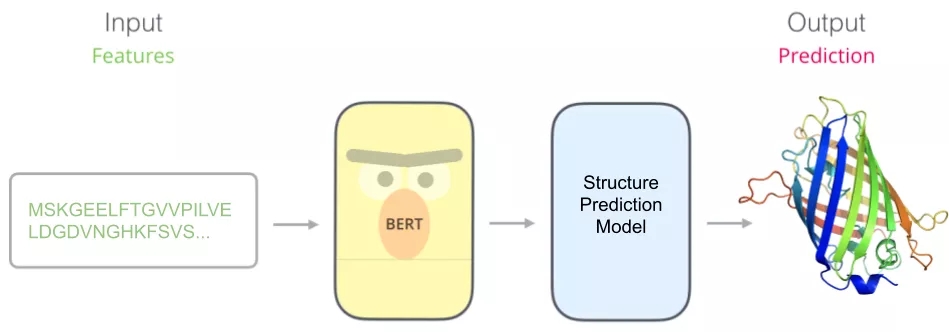
当我们得到预训练模型后,就可以将特定任务的模型堆叠在其顶部。同样的方法可以训练出蛋白质序列的预训练模型,并基于此进行结构预测。
为什么这种方法可以学习出蛋白质的结构特征呢?这主要源于语言学的假设:自然的蛋白质存在于一个可学习的流形上,这个流形在进化压力下十分倾向于复用多尺度的元件。通过观察大量的序列,模型可以学习到蛋白质如何构建的隐含过程,包括序列中的复用比例,不同位置间的交互以及进化出现的区域,这将提供巨大的帮助。
自监督学习为研究人员提供了一种在不同蛋白质族间实现信息迁移的有效方式,与比对方法相比,自监督模型可以充分利用其他蛋白质家族的部分信息来提供有效的特征预测。近年来很多研究组从多个角度对这一问题进行了尝试,为了评测这些不同的自监督方法对应蛋白质预测的效果,研究人员提出了一个称为TAPE尺度基准测试标准来对他们的优劣进行比较。
使用自监督学习预测有效的蛋白质结构
在生物学中绿色荧光蛋白(GFP)为研究人员提供了有效的生物标记方法,但人们一直想获得发光强度更高的荧光蛋白。如果对其分子结构进行改性,绝大多数都会造成蛋白质失活,只有极少部分可以保持蛋白质功能。如果可以知道哪些改变会造失活,就可以为研究人员提供更为明确的改性方向,大幅度提高蛋白质工程的效率。
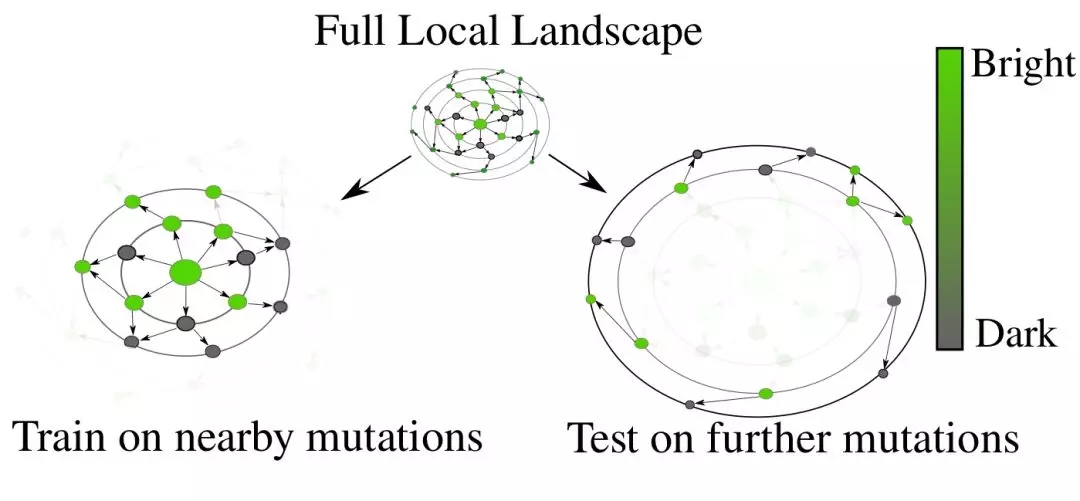
不同扰动下GFP分子的变化情况,与原始分子结构相距越远找到合适结构的概率就越小。
对于GFP来说,其每个位置有将近二十种可能的变换,那么仅仅五个位置的变化将会带来超过三百二十万种可能性,我们无法在实验室中合成那么多种结构。可行的方法是利用邻近的蛋白质变体训练模型,并通过目标函数来优化寻找更多可能的荧光变体。通过对这些可能的变体进行排序来指导实验室进行高效的合成实验。
在充分的训练后模型可以学习到蛋白质中每个氨基酸的嵌入,通过对这些嵌入矢量进行处理就可以得到变体的全局表示。下图展示了绿色荧光蛋白可能变体的分布,并用颜色表示的发光的可能性。
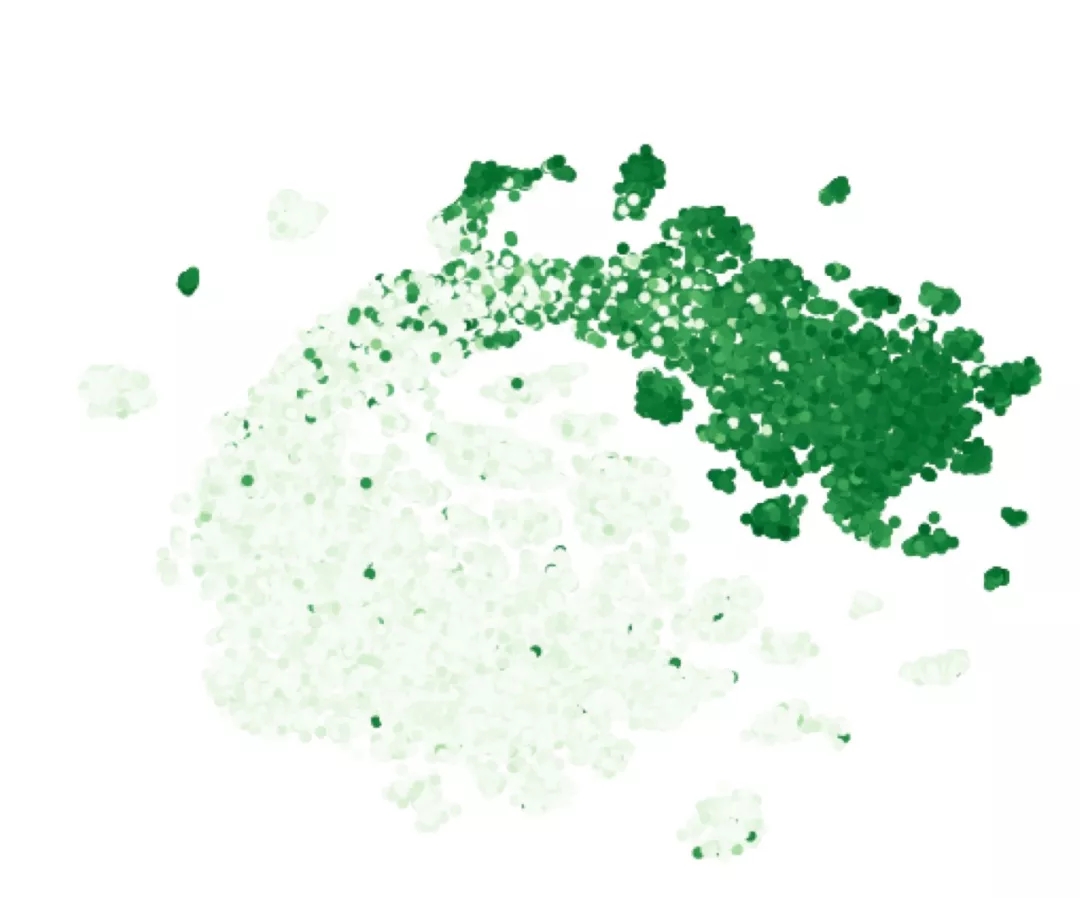
上图中清晰的显示了发光和不发光的变体,并且预测准确率相比原始方法提高了近五倍的水平。同时可以预测出每个位置的重要程度。通常低熵位置的变化会造成蛋白质的损坏,但本文提出的模型提供了别样的视角,除了熵值之外,不同位置间的稳定相关性也是蛋白质特性的重要度量。
除此之外,这一模型还可以用于蛋白质中序列的接触预测。
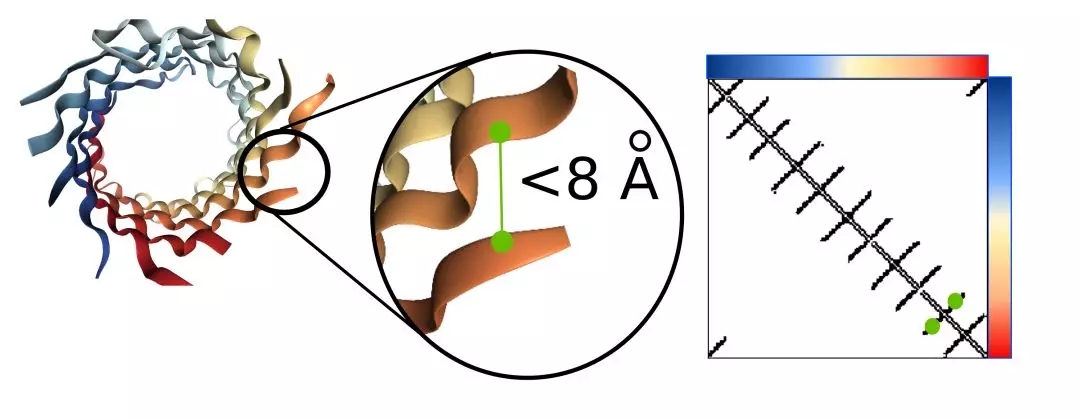
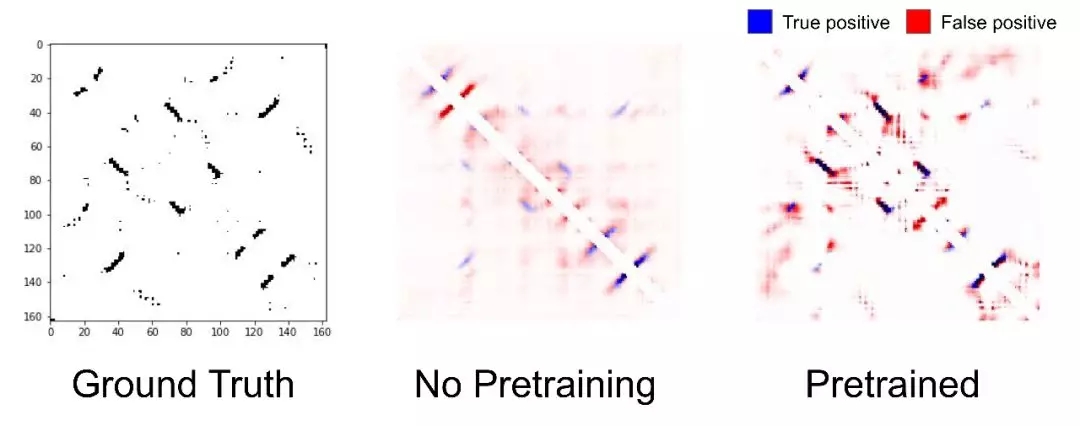
接触预测主要任务是预测某一对氨基酸是否接触(3D空间中距离小于<8Å)的0/1预测。精确的接触预测为三维结构建模提供了有效的全局信息。更为清晰的接触预测意味着更为可靠的预测。
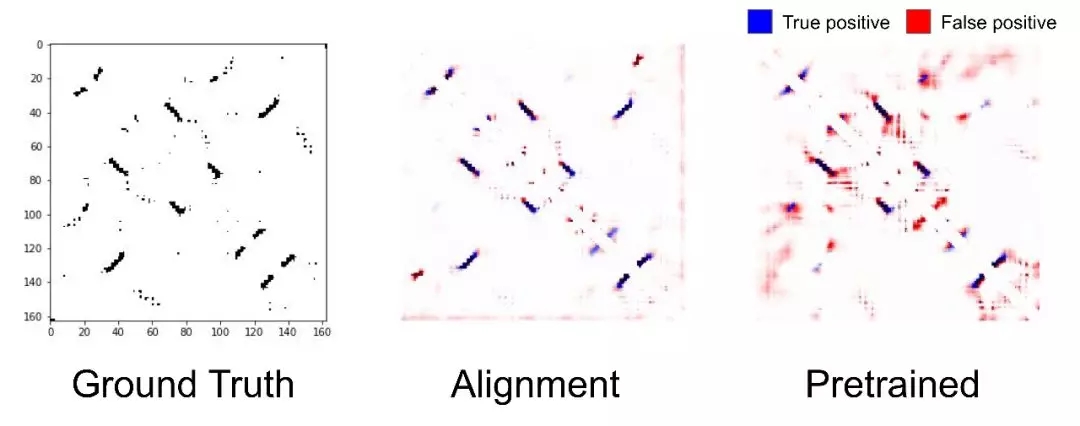
上图显示了基于预训练模型的蛋白质位置预测更为清晰。但需要指出的是,如果使用提前计算好的比对特征(下图中)作为输入,得到的结果由于利用LSTM(下图右)方法预测出的接触图,其中原因还有待进一步研究,但这也表明这一领域还有这巨大的提升空间。
机器学习对于蛋白质的有效预测是AI与生物有效结合的最佳例子之一,不仅仅会为世界带来十分深刻的影响,同时需要解决一系列实际挑战来促进机器学习本身的发展。蛋白质作为变长的长序列,同时会在三维空间上折叠变换,并于在不同氨基酸间发生相互作用,是一种十分复杂的数据形式。
随着生物学的发展,越来越多的蛋白质被采集、观察,机器学习应用于蛋白质的时机已经成熟,将会涌现出越来越多优秀的工作,让我们不断加深对于蛋白质这一生命原料的深刻理解和认识。
如果想了解更多细节,请参看论文TAPE,并上手尝试代码合成自己的新蛋白吧:
https://www.biorxiv.org/content/biorxiv/early/2019/06/20/676825.full.pdf
https://github.com/songlab-cal/tape









