合成机器人和 AI 联手发现高选择性催化剂 - IT思维
公众号/ ScienceAI(ID:Philosophyai)
编辑 | 绿萝

催化剂优化过程通常依赖于化学家基于筛选 数据 的归纳和定性假设。虽然使用分子特性或计算的 3D 结构的 机器学习 模型可以进行定量数据评估,但通常需要进行昂贵的量子化学计算。相比之下,现成的二进制指纹描述符具有时间和成本效益,但其预测性能仍然不足。
近日,来自日本北海道大学、德国马克斯·普朗克煤炭研究所(Max-Planck-Institut für Kohlenforschung)和法国斯特拉斯堡大学的研究团队,描述了一个基于片段描述符的机器学习模型,该模型针对不对称催化进行了微调,并表示环状或多环芳烃,从而实现稳健高效的虚拟筛选。使用仅具有中等选择性的训练数据,从理论上设计并通过实验验证了在具有挑战性的不对称四氢吡喃合成中表现出更高选择性的新催化剂。
该研究以「Predicting Highly Enantioselective Catalysts Using Tunable Fragment Descriptors」为题,于 2023 年 1 月 23 日发布在《Angewandte Chemie International Edition》上。

论文链接:https://doi.org/10.1002/anie.202218659
随着 ChatGPT 语言处理功能的出现,AI 最近成为头条新闻。为化学反应设计创建一个同样强大的工具仍然是一个重大挑战,尤其是对于复杂的催化反应。
「已经有一些先进的技术可以 [预测] 催化剂结构,但这些方法往往需要大量的计算资源和时间投入,但它们的准确性仍然有限,」共同第一作者 Nobuya Tsuji 说。「在这个项目中,我们开发了一个预测模型,你甚至可以在日常笔记本电脑上运行。」
为了让计算机学习化学信息,分子通常被表示为描述符的集合,这些描述符通常由这些分子的小部分或片段组成。这些对 AI 来说更容易处理,并且可以排列和重新排列来构建不同的分子,就像乐高积木可以以不同的方式排列和连接来构建不同的结构一样。
然而,计算成本更低的 2D 描述符一直难以准确表示复杂的催化剂结构,导致预测不准确。
为了改善这个问题,研究人员开发了新的圆形子结构 (CircuS) 二维描述符,明确表示催化中常见的环状和支链碳氢化合物结构。人工智能的训练数据是通过使用合成机器人的流线型半自动过程进行实验获得的。然后将该实验数据转换为描述符,并用于训练 AI 模型。
在此,研究人员基于 ISIDA 平台的预测模型,使用片段计数描述符来预测结构多样化且灵活的催化剂的对映体选择性,不需要任何量子化学计算。为了更精确地表示不对称催化中常见的环或多芳烃取代基,引入了另一种片段类型。此外,还展示了它在实际合成挑战中的应用;用于不对称 2,2-二取代四氢吡喃合成的高选择性催化剂已从仅显示中等选择性的催化剂组成的训练数据中预测出来。
研究人员对简单烯烃的立体选择性加氢烷氧基化反应进行了初步研究,以在几种不同条件下使用亚氨基双磷酰亚胺酸酯 (IDPi) 催化剂构建 2,2-二取代四氢呋喃环(35 个示例,e.r. 从 15:85 到 91:9;进一步参考为 THF 集)。这些 IDPi 催化剂具有共同的 scaffold,但在 BINOL 和氮取代基的 3,3′-位置上有多种取代基,表示为 Ar 和 R(为了简单起见,在 THF 组中只有 R=Tf)。
图 1B 总结了工作流程。催化剂、底物和产物的结构信息由指纹或片段描述符编码。在片段的情况下,反应方案被转化为 CGR。催化剂的结构可以表示为完整结构或仅由表示为 Ar 和 R 的可调取代基表示;后者将减少来自共同 scaffold 的噪音,并能够应用适合每个取代基的不同描述符。溶剂的物理化学参数也作为描述符包含在模型中。其他反应条件,即温度和浓度,分别考虑;在构建预测模型之前,所有数据点都被校准到相同的条件,并且预测的自由能差异被校准回每个实验条件,提供给定条件下的预测对映体比率。因此,该工作流程代表了一种同时处理催化剂、反应方案和反应条件的通用方法。

图 1:(A) 初始研究中使用的数据集 (THF)。(B) 一般建模工作流程。(来源:论文)
通过这些策略,进行了初步基准研究,以比较各种指纹和 ISIDA 片段计数的性能,其中考虑了原子中心片段和线性片段。由于数据集的大小适中,支持向量机(SVM)方法被选为广泛使用的非线性机器学习算法。描述符空间和模型超参数通过遗传算法在 10 折交叉验证(CV)中进行优化。在初步研究中,ISIDA 片段显示出比传统指纹更好的预测性。然而,仍然存在对其固有线性的担忧。

图 2:(A) 留一法 (LOO) 交叉验证中 THF 集的描述符基准测试结果。(B) CircuS 在距中心原子给定距离处生成的片段。(C) 最佳模型在 THF 数据集上的 LOO 交叉验证结果。(来源:论文)
为了解决这种复杂性,在 CGRtools 库的框架中开发了称为 CircuS(圆形子结构)的增强子结构类型描述符。它们的功能类似于以 ISIDA 原子为中心的片段,但明确考虑遇到的具有闭环的完整子结构,更具体地代表复杂的结构基序。正如预期的那样,CircuS 的表现甚至优于 ISIDA 描述符;基于催化剂取代基的 CircuS 片段构建的完全优化模型优于其他模型,在留一法 (LOO) 交叉验证中具有令人满意的 R^2=0.905、平均绝对误差 MAE=0.741 kJ/mol。
然后,研究人员设想应用已建立的协议(protocol)用于四氢吡喃 (THP) 合成的更具选择性的催化剂的计算机辅助设计。为了更有效的筛选和数据一致性,使用了合成机器人,简化了从实验到数据生成的过程。为了构建 THP 合成的预测模型,随后构建了包含 35 个 THF 示例和 35 个 THP 示例的通用数据集以确保催化剂结构的多样性。
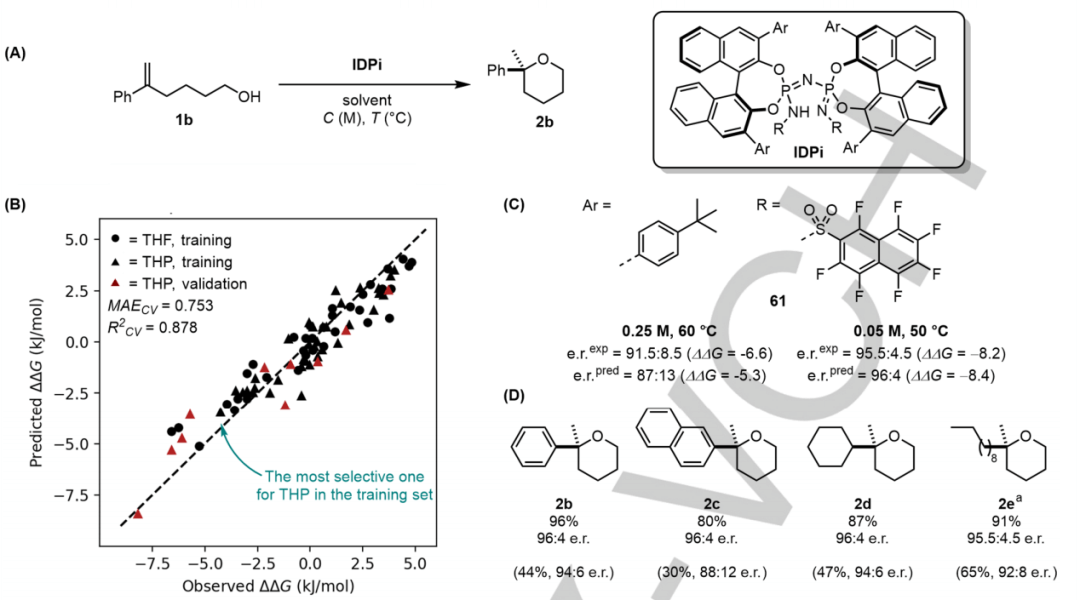
图 3:(A) 目标反应(THP 合成)以预测更具选择性的催化剂。(B) THF数据集和 THP 数据上最佳模型的10 倍交叉验证结果。(C) 用于比较不同反应条件下的实验值和预测值的选定示例。(D) 具有最佳催化剂 61 的底物范围。提供了单独的产量。(来源:论文)
基于 CircuS 片段描述符构建的完全优化模型提供了合理的预测性,R^2 CV=0.878,MAECV =0.754 kJ/mol。基于此,进行了虚拟筛选。
研究人员使用经过全面训练的模型虚拟测试了 190 种不属于训练数据的催化剂。在这组数据中,AI 模型仅在对具有中等选择性的催化剂数据进行训练后就能够预测高选择性催化剂,显示出超越训练数据进行推断的能力。然后对预测具有最高选择性的催化剂进行了实验测试,显示出与 AI 模型预测的几乎相同的选择性。
获得高选择性对于新药的设计尤为重要,这项技术为化学家提供了一个强大的框架来优化选择性,在计算和劳动力成本方面都是有效的。
为了测试开发的描述符的范围,研究人员的方案进一步应用于手性磷酸 (CPA) 催化的硫醇不对称加成到亚胺中的数据集。预测模型的性能,尤其是在一组外部催化剂上,比任何其他 2D 描述符更接近报告的 3D 描述符,这表明所描述方法的使用不限于特定反应。
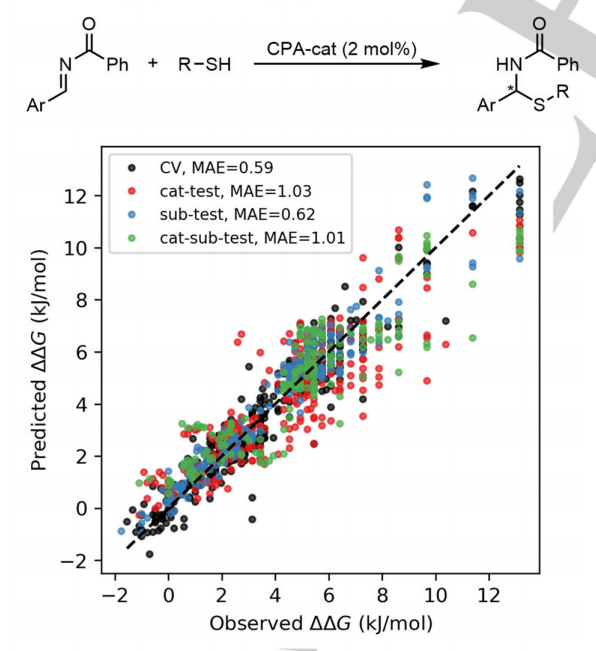
图 4:CPA 催化的硫醇不对称加成到以 CircuS 片段为代表的亚胺的 10 倍交叉验证 (CV) 和外部预测结果。(来源:论文)
「通常,为了预测新的选择性催化剂,化学家们会使用基于量子化学计算的模型。然而,这种模型的计算成本很高,当化合物的数量和分子的大小增加时,它们的应用就会受到限制,」共同第一作者 Pavel Sidorov 评论道。「基于 2D结构的模型要便宜得多,因此可以在几秒钟内处理成百上千个分子。这使得化学家可以更快地过滤掉他们可能不感兴趣的化合物。」
参考内容:https://phys.org/news/2023-02-robots-ai-team-highly-catalysts.html