神经网络学习预测蛋白「分子机器」的运动
公众号/ScienceAI(ID:Philosophyai)
编辑 | 橘子皮
来自美国莫格里奇研究所的 Timothy Grant 发表观点文章,评论《Nature Methods》上的两项蛋白质分子动力学方面的研究,并表示新的计算方法从冷冻电镜图像中捕获分子运动,并提供对蛋白质动力学的更完整理解。
该文章以「Neural networks learn the motions of molecular machines」为题,于 2021 年 7 月 29 日发布在《Nature Methods》。

温馨提示,干货在文章后半部分
虽然分子结构通常被视为静态和固定的,但实际上蛋白质及其复合物通常是极其动态的,表现出一系列运动,并且在任何给定点具有许多可能的三维排列。最近在 《Nature Methods》上发表的两项研究(一项由 Ellen D. Zhong 等人发表,另一项由 Muyuan Chen 和 Steven J. Ludtke 发表),已经证明了神经网络能够从给定分子的众多图像中学习多个结构,每个图像都冻结在不同的位置。然后,这些训练有素的网络能够生成一个结构集合,允许对样本中发生的动力学进行深入分析,最终导致对重要分子机制的更好理解。

Ellen D. Zhong 等人的论文。
论文链接:https://www.nature.com/articles/s41592-020-01049-4

Muyuan Chen 和 Steven J. Ludtke 的论文。
论文链接:https://www.nature.com/articles/s41592-021-01220-5
简单介绍背景
X 射线晶体学历来是解决高分辨率蛋白质结构的主要技术。该技术依赖于将分子形成的非常规则的重复晶体,因此晶体通常会将分子锁定为单一结构。过去人们倾向于认为分子结构是静态的、刚性的,但实际上这些结构往往是动态的。蛋白质内部发生的运动可能是其功能所固有的,因此它们可以被视为分子机器。
正如通过视频比从一张照片更容易理解汽车发动机的工作原理一样,拥有有关分子运动的详细信息,可以增强我们对这些蛋白质背后机制的理解,最终提高我们对它们的生物学理解,从而在药物设计时,增加对其功能进行修改编辑的可能性。
近年来,单粒子冷冻电子显微镜 (Cryo-EM) 已挑战 X 射线晶体学的主导技术称号,从仅生成低分辨率结构的方法转变为较大分子的首选结构技术。Cryo-EM 在解决的近原子分辨率结构的数量上,正在经历指数增长,并且最近在真正的原子级分辨率下进行了第一次观察,已经能够准确定位单个原子。冷冻电镜的工作原理是,使用电子显微镜对嵌入薄冰层中的分子进行成像。
用于对样品成像的高能电子也会迅速破坏它们,因此必须使用很少的电子对样品进行成像——就像在黑暗中拍照一样。这会导致图片背景非常嘈杂,从而使图片难以解释。然而,如果利用冷冻电镜实验记录目标分子的数千个不同副本的图像,将这些单独的图片计算组合,便有可能得到分子的真实结构。
该技术能够对处于溶液状态的分子进行成像,因此来自冷冻电镜实验的数据将包含不同位置阵列中分子的各种快照。这既是一种祝福也是一种诅咒:一方面,它使求解结构变得更加复杂,并且在非常大的运动的情况下,可能使求解实际上变得不可能。另一方面,如果这个问题能够得到解决,单个冷冻电镜实验就可以揭示大量信息,不再只提供单一的静态结构,而是提供一系列结构和对机制的更深入了解(如下图)。
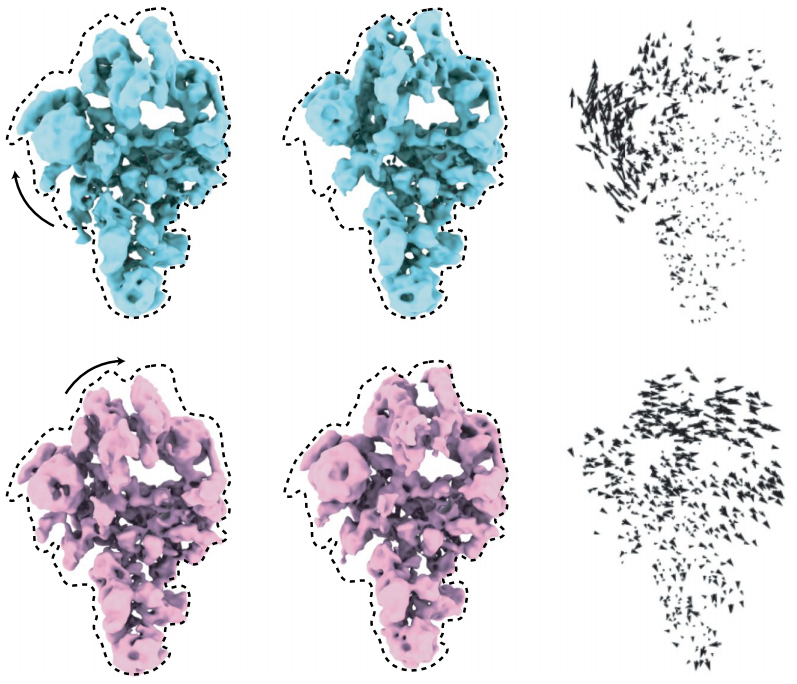
酵母剪接体的两种可能的运动,一个高度动态的复合体。
介绍方法
Ellen D. Zhong 和 Muyuan Chen 等人使用深度学习来解决这个问题;创建神经网络,当呈现不同位置的数千张分子图像时,能够学习潜在的结构,包括任何存在的运动。这两种方法都通过让神经网络学习在抽象的「潜在空间」中对粒子图像进行编码,基本上只使用几个数字来描述整个图像。与此同时,另一个神经网络学习如何将这几个数字转换或解码为分子的三维结构。
结合这两个网络,不仅学习编码和解码分子的初始静态结构,还学习编码和解码结构变化——例如,由于潜在运动引起的变化。这两种方法的另一个关键特征是,通过检查从所有分子图像创建的学习抽象空间,可以看到对应于不同结构的不同区域和模式,还可以展示它们之间的结构路径。
两种方法的不同
尽管这两种方法在使用深度学习来学习结构的集成方面相似,但它们在结构的表示方式上有所不同。Ellen D. Zhong 等人的研究,将表示保持为图像,网络学习从图像编码并解码为完整体积,直接将观察到的差异与体积联系起来。相比之下,Muyuan Chen 将体积表示为高斯分布的组合——球体加在一起形成结构的近似值——网络从图像编码,然后解码成具有不同位置和大小的球体列表。这简化了它们的表示,允许更容易地跟踪特定运动,同时允许关注特定区域或类型的变化;但在某些情况下可能会导致错误 ——例如,高度移动的区域可能永远不会被球体填充或可能使用的球体太少,很难准确描述小细节和小差异。
展望
目前,这两种方法都需要对结构进行初始估计,要求使用传统方法至少已经解决了其中的一部分。未来,一个令人兴奋的前景是网络有可能在没有任何先验信息的情况下,学习给定样本中的潜在结构和运动,从而可能解锁当前方法无法获得的结果。
即使在目前的形式中,这两项研究的解决方案,表明冷冻电镜终于开始发挥其全部潜力,揭示蛋白质的真正动态性质。对于该领域来说,这是一个激动人心的时刻,它可能会改变我们对蛋白质外观的看法,从漂浮在太空中的一个静态原子到一个动态机器,优雅地操纵以完成它们的任务。
论文链接:https://www.nature.com/articles/s41592-021-01235-y