公众号/
编辑 | 萝卜皮
开发用于在分子和纳米尺度上分析蛋白质-蛋白质相互作用(PPI)的新方法可以深入了解细胞内信号通路,并将提高对蛋白质功能以及其他生物和非生物来源的纳米级结构的理解。计算工具的最新进展,特别是涉及现代深度学习算法的工具,已被证明可以补充描述和合理化 PPI 的实验方法。
然而,现有的大多数 PPI 预测工作都使用蛋白质序列信息,因此难以解释蛋白质链的三维组织。
在近期的一项研究中,印度塔塔咨询服务公司与美国密歇根大学的研究人员共同解决了这个问题,并描述了一种基于图注意网络的 PPI 分析,称为 Struct2Graph,用于直接从折叠蛋白球的结构数据中识别 PPI。该方法能够在由相等数量的正负对组成的平衡集上以 98.89% 的准确度预测 PPI。在正负对比例为 1:10 的不平衡集上,Struct2Graph 实现了 99.42% 的五倍交叉验证平均准确率。
此外,Struct2Graph 可以潜在地识别可能有助于形成蛋白质-蛋白质复合物的残基。针对两种不同的相互作用类型测试重要残基的鉴定:(a)具有多个配体竞争相同结合区域的蛋白质,(b)动态蛋白质-蛋白质粘附相互作用。Struct2Graph 以 30% 的灵敏度、89% 的特异性和 87% 的准确度识别相互作用的残基。
该研究以「Struct2Graph: a graph attention network for structure based predictions of protein–protein interactions」为题,于 2022 年 9 月 10 日发布于《BMC Bioinformatics》。

蛋白质-蛋白质相互作用(PPI)是许多生物过程的基础。对人类蛋白质组的分析表明,大多数蛋白质并非单独发挥作用,而是作为多单元复合物的一部分。事实上,PPI 是信号转导、代谢调节、环境感知和细胞组织的核心部分。
在这些过程中,PPI 可以改变酶动力学、促进底物通道、形成新的结合位点、使蛋白质失活或改变蛋白质相对于底物的特异性。由于 PPI 在生命系统中无处不在,能够表征这些相互作用有望进一步了解细胞过程,并为疾病治疗和药物发现提供不可或缺的工具。PPI 及其数学描述对于从其他纳米级构建块(包括但不限于脂质、糖、聚合物、纳米级缀合物和无机纳米粒子)创建蛋白质类似物也是必不可少的。学界已采用许多策略来解码主要针对分子尺度数据和氨基酸序列的 PPI。
高通量实验技术如双杂交筛选、串联亲和纯化和质谱已被用于创建蛋白质相互作用网络。然而,对这些传统方法准确性不足、实验吞吐量低和成本高的担忧,激发了对可补充传统和机器人实验协议的计算方法的研发。计算方法可以根据蛋白质的遗传背景、氨基酸序列或结构信息的数据来预测蛋白质是否会相互作用。在确定一对蛋白质是否相互作用时,基因组学分析会考虑基因融合、常见物种之间的保护(系统发育分析)和进化历史等因素。
PPI 分析的典型计算技术使用两种蛋白质的氨基酸序列来确定是否发生相互作用。已经提出了许多特征,例如公共子序列的频率和自协方差,以将不同长度的序列转换为统一大小的表示。基于序列的方法最近能够利用蛋白质数据库和机器学习技术进行高精度预测。
来自序列的蛋白质-蛋白质复合物的三维(3D)结构可以通过 CO-threading 算法(COTH)预测,该算法从已解决的复杂结构数据库中识别蛋白质复合物的模板。COTH 使用评分功能和结构信息比对氨基酸链序列。DeepPPI 模型使用人工神经网络预测交互,该网络将捕获序列的组成、分布和顺序的特征向量作为输入。DeepFE 对氨基酸序列使用自然语言处理算法来创建适合作为神经网络分析输入的序列的低维嵌入。尤其是 DeepFE,已被证明非常有效,在酿酒酵母和人类数据集上的预测准确率分别为 94.78% 和 98.77%。
事实上,大多数基于深度学习的方法已被证明可以实现高 PPI 预测精度,因为它们具有更大的表示能力。除了纯粹依赖基于序列的信息外,现代机器学习方法通常还结合网络级信息来进行 PPI 预测。在 PPI 网络中,每个节点代表一个蛋白质,而它们之间的边代表相互作用。因此,预测任何两个节点之间的交互是一个变相的链接预测问题。
最近,有些方法利用网络结构以及使用氨基酸序列的矢量化表示来获得更强的预测性能。尽管取得了成功,但上述基于序列的方法并未推广到与蛋白质相似规模的更广泛类别的化合物,这些化合物同样能够与不基于氨基酸的蛋白质形成复合物,因此缺乏基于序列的等效表示 。
虽然可以准确预测蛋白质与 DNA 的相互作用,但基于机器学习的预测高分子量脂质、糖、聚合物、树枝状聚合物和无机 纳米 颗粒的蛋白质复合物的方法在纳米医学和纳米诊断学中受到广泛关注,但在实验人员中并不广为人知,尽管随着蛋白质和纳米颗粒的统一结构描述符的发展,这一方向取得了重大进展。
因此,考虑到蛋白质结构及其可变的非蛋白质、仿生和非生物对应物的预测计算方法成为可能。一些方法使用蛋白质的 3D 结构预测相互作用,使用基于知识的方法来评估候选蛋白质与模板蛋白质复合物的结构相似性。由于这种方法需要有关更大复杂的详细信息,无模板对接方法分析未结合的蛋白质成分,并从大量潜在的相互作用位点中识别出最有希望的相互作用。虽然对接方法已经显示出对某些蛋白质的成功,但它们面临着蛋白质在相互作用过程中发生构象变化的困难。许多这些结构方法也作为机器学习模型的基础。
2012 年,Zhang QC 团队开发了 PrePPI,它使用氨基酸序列和系统发育特征作为朴素贝叶斯分类器的输入。2018 年 Northey TC 团队开发了 IntPred,它将蛋白质分割成一组补丁,将 3D 结构信息整合到一个特征集中,以预测与多层感知网络的交互。这些模型在精心策划的交互数据库上进行训练,描述蛋白质之间的二元相互作用以及相应的接口位点或原子。
在最近的一项工作中,印度塔塔咨询服务公司与美国密歇根大学的研究人员迈出了评估蛋白质与其他纳米结构的超分子相互作用的通用方法的第一步。与氨基酸氨基酸序列信息相比,所提出的方法从晶体学数据确定蛋白质纳米级表示中蛋白质-蛋白质复合物形成的概率。
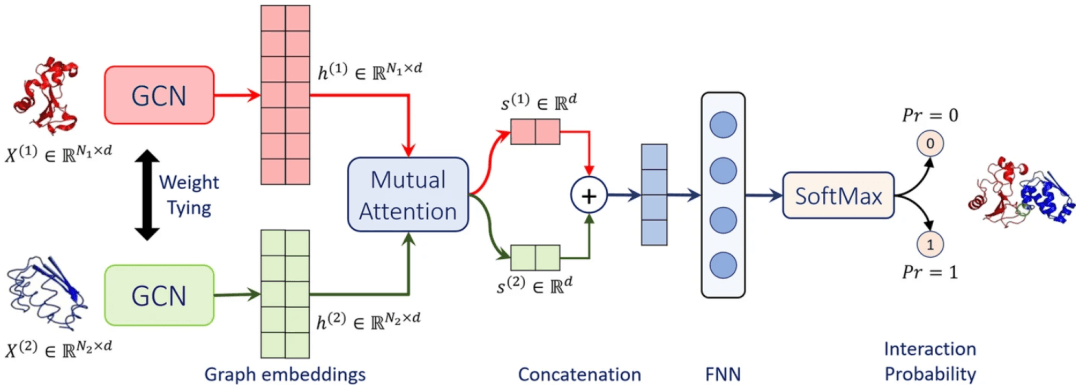
图示:Struct2Graph 架构示意图。(来源:论文)
该团队开发了一个相互图注意力网络和一个相应的计算工具 Struct2Graph,以仅从 3D 结构信息预测 PPI。Struct2Graph 没有使用几个蛋白质特定的特征,例如疏水性、溶剂可及表面积(SASA)、电荷、ngram 频率等,而是使用仅使用原子的 3D 位置获得的基于图形的蛋白质球表示。这种基于图的解释允许神经信息传递,以实现蛋白质的有效表示学习。
Struct2Graph 建立在该团队之前关于代谢途径预测工作的基础上,该工作表明,小分子和肽的等效基于图的结构表示与图卷积网络相结合,显著优于其他涉及计算各种生化特征作为输入的分类器。这种方法还利用图论的泛化来描述类似于 PPI 的复杂纳米级组件。
该方法能够在由相等数量的正负对组成的平衡集上以 98.89% 的准确度正确预测 PPI。在正负对比例为 1:10 的不平衡集上,Struct2Graph 实现了 99.42% 的五倍交叉验证平均准确率。Struct2Graph 不仅优于经典的基于特征的机器学习方法,而且优于其他现代深度学习方法,例如使用序列信息和特征选择进行 PPI 预测的 Deep-PPI 和 DeepFE-PPI。
除了 PPI 预测的高精度之外,Struct2Graph 还提供了许多优势。与利用几何仿生学思想的 ML 算法类似,Struct2Graph 只需要单个蛋白质的 3D 结构。
另外,虽然这里研究人员专注于蛋白质相互作用,但通过在他们的分析中仅使用原子的位置,该框架可以推广到其他可用 3D 信息的分子结构。此外,Struct2Graph 还能够深入了解蛋白质相互作用的性质。通过其注意机制,该模型可以潜在地识别可能有助于形成蛋白质-蛋白质复合物的残基。与其他模型不同,Struct2Graph 能够以无监督的方式生成这些数据,因此不需要通常无法获得的蛋白质复合物信息。
重要残基的鉴定针对两种不同的相互作用类型(训练集的一部分)进行测试:(a)具有多个配体竞争相同结合区域的蛋白质,(b)动态蛋白质 – 蛋白质粘附相互作用。Struct2Graph 以 30% 的灵敏度、89% 的特异性和 87% 的准确度识别相互作用的残基。
另外,在已知的 2724 个致病 SAV 和 1364 个多态性中,该团队的注意力机制将所有致病 SAV 中的 33.55% 确定为重要的(注意力权重在前 20% 以内),而 85.30% 的多态性被提议的注意机制确定为不重要,表明该团队先前建立的 SAV 研究与提议的注意机制确定的重要残基之间存在显著重叠。
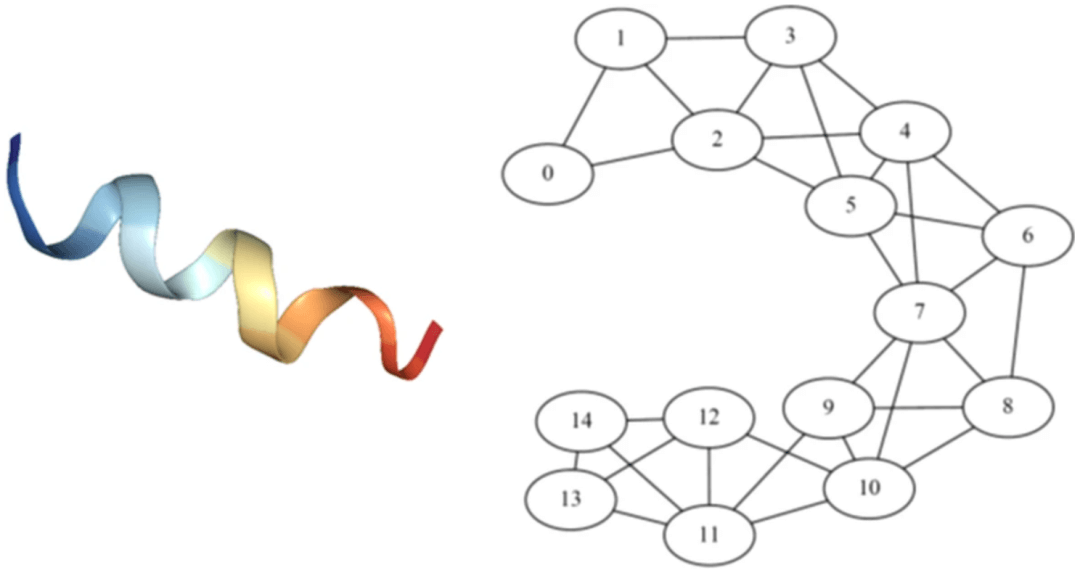
图示:蛋白质和蛋白质图。(来源:论文)
总之,该团队使用第一个基于 3D 结构的图形注意网络来解决 PPI 预测问题。新颖的相互注意机制通过其无监督的知识选择过程提供了对可能交互站点的洞察。研究表明,从单个蛋白质的图结构中学习到的相对低维的特征嵌入优于其他基于全局蛋白质特征的现代机器学习分类器。另外,通过对单个氨基酸变异的分析,注意力机制显示出对致病残基变异的偏好优于良性多态性,表明它不仅限于界面残基。
开源地址:https://github.com/baranwa2/Struct2Graph
论文链接:https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-022-04910-9