公众号/
编辑 | 萝卜皮
化合物效力预测是机器学习在药物发现中的一种流行应用,为此使用了越来越复杂的模型。总体目标是识别对给定目标高度有效的新化学实体。效力预测模型的相对性能及其准确性限制在该领域继续存在争议,深度学习是否可以进一步推进效力预测仍不清楚。
波恩大学(Rheinische Friedrich-Wilhelms-Universität Bonn)的研究人员分析和比较了不同计算复杂度的效力预测方法,并表明简单的最近邻分析(Simple nearest-neighbour analysis)被视为该领域最先进的机器学习方法的准确性标准。此外,使用不同模型的完全随机预测被证明可以在一个数量级内重现实验值,这是由常用化合物数据集中的效力值分布产生的。总而言之,这些发现对于评估机器学习性能的典型基准计算具有重要意义。模型评估中通常应包括最近邻分析等简单控制。此外,最好的和完全随机的效力预测之间的狭窄界限是不现实的,需要考虑替代基准标准。
该研究以「Simple nearest-neighbour analysis meets the accuracy of compound potency predictions using complex machine learning models」为题 于 2022 年 12 月 14 日发布在《Nature Machine Intelligence》。

在化学信息学和药物化学中,化合物效力或其他分子特性的预测起着核心作用。对于效力预测,应用了基于配体和结构的方法,其中许多方法采用机器学习 (ML)。与人工智能已成为焦点的其他领域一样,在基于结构和基于配体的建模中,复杂的深度学习架构越来越多地用于效力/特性预测。然而,尽管取得了明显的进步,但其中一些预测也存在争议。
基于配体的效力预测解释非线性结构-活性关系是化学信息学的支柱。为此,受监督的 ML 模型是在一组已知活性化合物的基础上推导出来的,以预测新分子的效力。虽然识别新型活性化合物的前瞻性应用代表了最终目标,但模型性能最初是通过基准测试进行评估的,这是报告新计算方法的先决条件。在典型的基准设置中,针对特定目标(通常称为活动类别)具有活动的复合数据集被分为训练和测试集,并使用交叉验证协议评估预测。在报告新方法和预测模型时,通常会遵循这条路线。
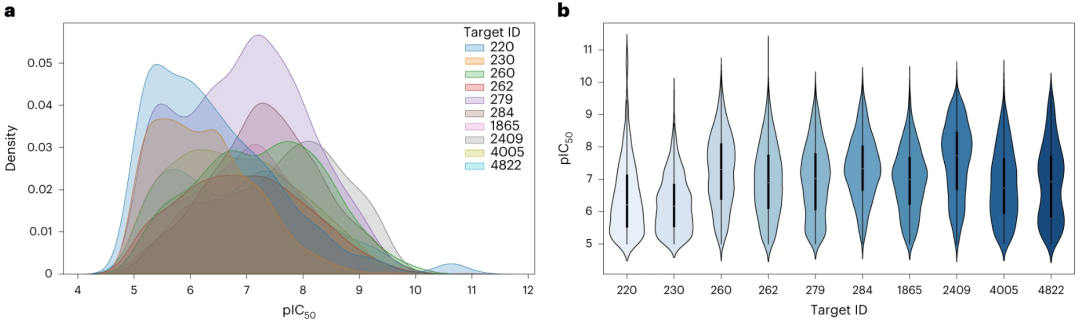
图:活动类别的效能值分布。(来源:论文)
在波恩大学研究团队开发用于识别新型活性化合物和预测其效力的计算方法的过程中,研究人员已经研究了数百个活性类别,方法的复杂性差异很大。在这些研究中,研究人员经常观察到方法论的复杂性与预测准确性无关。这里,研究人员报告了具有代表性的效力预测,证明简单的 k 最近邻 (kNN) 方法始终达到或超过高级 ML 方法的准确性,包括支持向量回归 (SVR),该领域广泛应用的标准,随机森林回归 (RFR)、深度神经网络 (DNN) 和具有表示学习的图卷积神经网络 (GCN)。DNN 和 GCN 代表了越来越流行的用于分子效力/特性预测的深度学习方法。
该研究的结果指出了目前很少考虑的化合物效力预测中的两个关键问题。首先,与简单的 kNN 计算相比,使用复杂的 ML 模型进行效力预测几乎没有任何好处。其次,性能最佳的 ML 模型和完全随机预测的区别仅在于较小的 MAE 裕度,对应于相对于实验观察的效力差异不到十倍。这两个问题都需要进一步考虑。
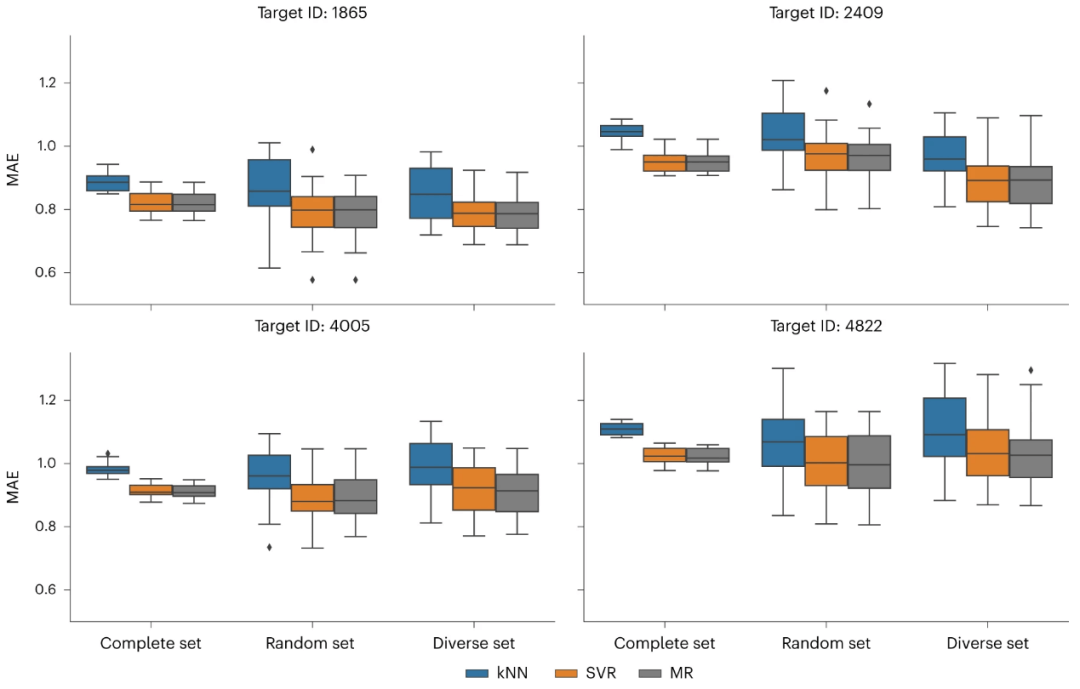
图:随机预测模型的性能。(来源:论文)
kNN 分析以前已成功用于化学相似性搜索和化合物分类。基本原理在药物化学中很常见:许多相似的化合物(例如结构类似物)具有相似的效力。值得注意的例外是活动悬崖(即具有较大效力差异的结构类似物)。然而,由于只有约 5% 的生物活性化合物参与了不同活动类别的活动悬崖的形成,因此它们对基于统计模型的效力预测准确性的影响在很大程度上可以忽略不计。对于以微纳摩尔到高纳摩尔范围内的化合物为主的大型数据集尤其如此,这通常是可用于基准测试的活动类别的情况。显然,诸如 kNN 计算之类的简单方法通常应用作评估复合效力/特性预测的新计算方法的参考。这里呈现的结果表明,可能很难确定 ML 方法优于这些简单预测的优势。对于尺寸缩减和结构多样化的训练集,kNN 与 SVR 相媲美,并且表现优于 DNN/GCN,尽管使用了大量化合物进行学习。即使对于保留组的结构独特或最有效的化合物,对于不同复杂性的效力预测模型也观察到总体相似的性能。
此外,最佳预测和随机预测之间的微小差距揭示了传统基准测试的一个主要缺点。简单地将训练集的中值效力值分配给任何测试化合物 (MR) 会产生 0.8–1.0 的 MAE。值得注意的是,这个误差范围与完全随机的预测非常匹配。这些发现是来自药物化学的活性类别的效力值分布的直接结果。在实际应用中,在一个数量级(十倍)内持续预测新化合物的效力将是相当成功的。然而,在基准设置中,随机预测人为地产生了这种水平的「伪准确性」。因此,在这些条件下,即使不是不可能,也很难评估计算方法的「真实」性能。
因此,研究人员可能会优先关注未来的应用,以预测和实验验证新化合物的效力。然而,在虚拟筛选文献中,一个常见的误解是,如果识别出一种或多种新的活性化合物,则计算方法是「有效的」。然而,情况并非如此,除非最终证明更简单的方法不能识别相同或相似的化合物。在前瞻性效力预测中,这也需要使用参考方法,例如 kNN。
还有其他具有挑战性的预测任务需要特别考虑。例如,来自药物化学的后期先导优化数据通常包含许多非常相似的化合物,具有可比(通常相对较高)的效力,并且只有少数代表活动悬崖的「异常值」。从统计的角度来看,这种结构-效力关系的普遍存在也有利于简单的 kNN 预测,但它们不适用于搜索形成活动悬崖的最有趣的类似物。这为能够定量解释结构-效能关系中高度不连续性的统计不足实例的方法留下了很大空间。
总之,根据此处报告的发现,很明显,计算效能预测的基准设置和复杂 ML 模型的表观性能需要重新评估。向前迈出的一步可能是将方法学评估的重点放在预测包含模型推导过程中未考虑的结构独特且高效化合物的保留集上。这至少会减轻由基准数据集中的全局效力值分布和复合相似关系引起的一些限制,并解决计算效力预测的最重要的实际目标。此外,基准数据集可能被设计为用来自不同系列的化合物同等地填充装箱效力区间。结合使用 kNN 作为一般参考方法,更有意义的基准设置将有助于避免高估效力预测模型,并对其实际应用潜力提供更现实的评估。
论文链接:https://www.nature.com/articles/s42256-022-00581-6